
Leukemia
Introduction
Hello, welcome to this 12th class of the Hematology course provided by the University of Guanajuato. In this class we are going to review some basic concepts of acute leukemia. They are a group of disorders characterized by the accumulation of abnormal white blood cells in the bone marrow. These abnormal cells can cause bone marrow failure, a high circulating white blood cell count, and can infiltrate into other organs.
Common manifestations, although not essential, include:
- Abnormal leukocytes in peripheral blood.
- Elevated total leukocyte count.
- Evidence of bone marrow failure (for example, anemia, neutropenia, thrombocytopenia).
- Involvement of other organs (eg, liver, spleen, lymph nodes, meninges, brain, skin, or testicles).
These entities evolve quickly, and the patients worsen in a matter of months. Diagnostic features proposed by WHO are going to be reviewed, as well as some treatment options either for myeloid or lymphoid acute leukemias.
Content development
Acute leukemia is a group of blood disorders conformed mainly by malignant cells. The classification will be made according to the predominant cell, myeloid or lymphoid.
Etiology
It is unknown. It may be due to the interaction of various factors.
l. Chronic bone marrow disorders
Polycythemia vera
Myelosclerosis
Aplastic anemia
Myelodysplastic syndromes
Clonal expansion caused by somatic mutation of a single medullary, peripheral lymphoid or thymic cell.


II. Infection.
Leukemia in mice and birds can be transmitted by cell-free filtrates.
In humans, there is strong evidence of a viral etiology (leukemia / T-cell lymphoma).
Human T leukemia virus (HTLV), a RNA-type retrovirus, has been detected by electron microscopy and culture in the cells of patients with a particular type of T-cell leukemia / lymphoma, which is common in certain provinces of Japan. and that occurs sporadically elsewhere (black people from the Caribbean and the United States).
III. Radiation.
There is a higher incidence of leukemia in survivors of the atomic bombings in Japan, in patients with ankylosing spondylitis who have received spinal radiation, and in children whose mothers received abdominal radiation during their pregnancy.
IV. Hereditary.
There are reports of several cases occurring in a family and in identical twins.
There is a fairly high incidence of leukemia in some inherited diseases, especially Down syndrome, Fanconi anemia, Bloom syndrome, and ataxia telangiectasia.
V. Chemical agents.
Chronic exposure to benzene (bone marrow dysplasia and chromosomal changes) rare.
Chemotherapeutic agents: alkylating agents (chlorambucil, nitrogen mustard, melphalan, procarbazine).
Classification
A classification of leukemias is based on how quickly the disease develops and worsens:
ACUTE: usually develops rapidly. The number of leukemic cells increases rapidly, and these abnormal cells do not have a proper function. People with leukemia feel very tired, bruise easily, and suffer from frequent infections.
CHRONIC: usually develops slowly. Leukemic cells work almost the same as normal cells. People may not feel sick at first, and the first sign of illness may be abnormal results on a routine test. If left untreated, the leukemia cells will invade the bone marrow, crowding out the normal cells.
Acute leukemia
Malignant clonal proliferations of blast-type immature hematopoietic cells that can affect bone marrow, peripheral blood, or other tissues.
The diagnosis is based on the observation of a medullary blastosis that equals or exceeds 30% of the total cell, or 20% according to the recommendations of the World Health Organization (WHO), since the finding of some blast cells, both in bone marrow and in peripheral blood, it does not necessarily indicate the existence of acute leukemia.

% Blasts should be obtained, where possible, from a differential leukocyte count of 200 cells from a blood smear and 500 cells from a bone marrow aspirate, stained with Wright-Giemsa.
Determination of CD34-positive blasts by flow cytometry is not recommended as a substitute for visual inspection, since not all leukemic blasts express it.
Multiparameter flow cytometry should be used to determine blast lineage and detect aberrant antigenic profiles.

Leukemias are also classified by the type of white blood cell that is affected:
MYELOID: Leukemia begins in myeloid cells (megakaryocytic, granulomonocytic, or erythroid), and is called myeloid, myelogenous, or myeloblastic. Leukemia cells can be obtained from the bone marrow or from the blood.
LYMPHOID: Leukemia begins in lymphoid cells and is called lymphoid, lymphoblastic, or lymphocytic. Lymphoid leukemic cells can be obtained from bone marrow, blood, and lymph nodes, which can become inflamed.
Mixed leukemias: express markers of both lines
Undifferentiated leukemias: proliferating hematopoietic series cannot be determined.
Leukemia identification
MORPHOLOGY
Number of blasts
Myeloid vs lymphoid
Canes of Auer
CYTOCHEMISTRY
Pillar of the FAB classification, not used very frequently in WHO
Myeloid series: myeloperoxidase, Sudan Black B
Monocytic series: Non-specific esterase
GENETICS
Indicates the prognosis of patients
FLOW CYTOMETRY
Confirm the presence of blasts with specific markers
Detection of immunophenotypic aberrations
NOT a substitute for morphological examination.
Cytochemical techniques
The cellular components that can be revealed with the cytochemical study are:
Enzymes (oxidants and hydrolytics)
Glycogen
Lipids
The most common cytochemical stains are:
Myeloperoxidase staining
Granulocyte Alkaline Phosphatase (FAL or FAG) stain
Acid Phosphatase stain (FAC, lymphocytes)
Sudan Black B (lipids)
Esterases
Periodic acid Schiff (PAS) stain (glycogen)

Overview of the myeloid alterations

Classification of the fab. Acute myeloid leukemia
They divide acute myeloid leukemia into 8 subtypes, based on the type of cell the leukemia developed from and how mature they are. Although it is still used, it is not considered an updated classification, since it does not consider molecular alterations.

MPO: myeloperoxidase. SBB: Sudan Black B (Myeloblasts). NSE: Non-specific esterase (monocytes).


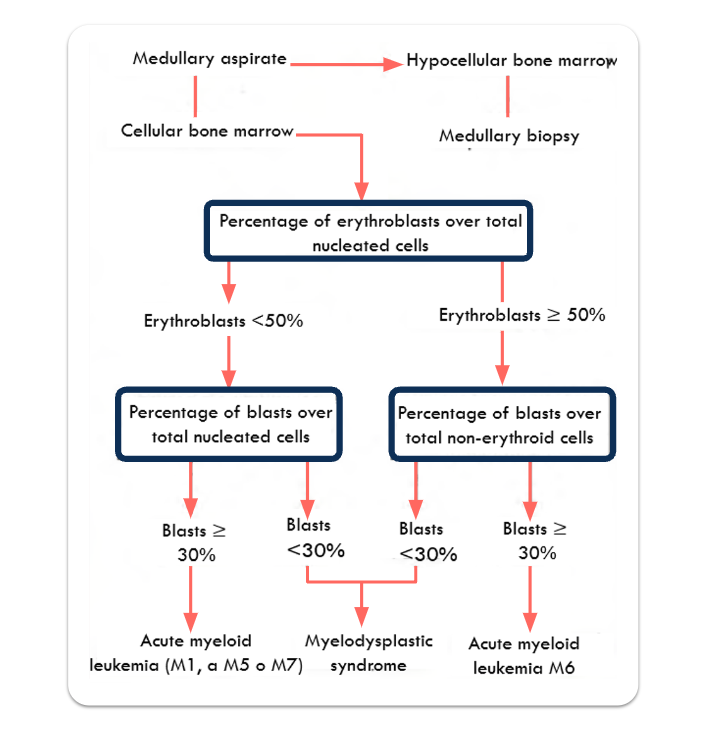
Procedure to follow in the analysis of a marrow aspirate to differentiate an acute myeloid leukemia from a myelodysplastic syndrome.
Who classification
It considers a value of >20% blasts in bone marrow or peripheral blood.
It considers cytogenetics and molecular lesions.
It considers previous illnesses and treatments.
The use of 2 or 3 antibodies for each of the three main lines -B, T and myeloid- allows to establish an immunological classification of acute leukemias, which correctly identifies the lineage in 98-99% of cases.




NPM1 is associated with nucleolar ribonucleoprotein structures and binds single-stranded nucleic acids
RUNX1-RUNX1T1: appears to be associated with the nuclear corepressor / histone deacetylase complex, thereby blocking hematopoietic differentiation.
FLT3: receptor tyrosine kinase with activity in the survival and proliferation of hematopoietic stem cells
CEBPA: interacts with the cyclins Cdk2 and Cdk4, inhibiting their kinase activity and thus preventing the growth of cells in culture.

Relatively Specific genetic markers
M2 t(8;21) (q22;q22)
M3 t(15;17) (q22;q21)
M4 inv(16) (q13;q22) o del((16) (q22)
M4 (M5) t(8;16) (p11;p13)
M5 t(9;11) (p21;p23)
Others
t(9;22) (q34;q11)
t(6;9) (p23;q34)
t(3;3) (q21;q29) inv(3) (q21q26)
+8
+21
5q-/-5
7q-/-7
t o del 12p11-p13

Very unfavorable prognosis: monosomies on chromosomes 5 and 7, deletions of the long arm of chromosome 5.
Monosomic karyotype: worse prognosis; two autosomal monosomies or one monosomy plus one structural abnormality.
Clinical prognostic markers:
Age
Patients older than 60 years are more likely to present unfavorable chromosomal abnormalities as well as to present comorbid medical conditions that make the use of intense chemotherapies difficult. They are also more likely to suffer from AML secondary to previous MDS, which confers a worse prognosis.
Leukocyte count
A high number (> 100,000) at the time of diagnosis is related to a worse prognosis.
Previous blood disorders or cancers
Previous hematological disorders (polycythemia vera, Fanconi anemia, congenital neutropenia, etc.) as well as myelodysplastic syndromes, are related to a poor prognosis of AML.
Treatment-related AML
Clinical manifestations of acute myeloblastic leukemias
They occur due to spinal cord failure, due to leukemic proliferation and infiltration of various organs and tissues.
80% of patients present anemia of variable intensity, of the normocytic-normochromic type.
Thrombocytopenia is recorded in 80-90% of cases, with 40-60% of patients showing hemorrhagic signs at the time of diagnosis, more frequent if the platelet count is less than 20 X 109 / L.
Fever is also a frequent clinical feature and is mainly due to an infectious etiology. Between 30 and 50% of patients present neutropenia, and it is the maximum cause of infections.
Multiple and nonspecific manifestations (3 months)
Presentation:
- Fatigue (1st symptom in 50%),
- Infections / fever (10%)
- Hemorrhage (5%)
- Bone pain; adenopathy; cough; headaches or sweating
- Paleness, dyspnea on exertion, and fatigue.
Infiltration
- Liver → hepatomegaly
- Spleen → splenomegaly
- Skin → leukemia cutis
- Lymph nodes → lymphadenopathy
- Bones → pain
- Gingiva, CNS, etc…
Promyelocytic leukemia
Its effectiveness increases with chemotherapy, It is frequent in Mexico and it is the one who has the best prognosis.

Treatment becomes an emergency when intravascular coagulation is prominent, causing a high proportion of early deaths (10-40%), mainly due to cerebral hemorrhages.
Differential therapy using all-trans retinoic acid (ATRA), combined with cytotoxic arsenic trioxide chemotherapy, has achieved a complete remission of 90%. It is the only cancer that to date has been treated with differential therapy.
The prognosis of APL, treated with ATRA and anthracycline, is more favorable than any other AML, and cases of relapse or refractory APL generally show a good response with arsenic trioxide.
DC56 expression is associated with a less favorable prognosis, whereas FLT3-ITD mutations are unclear.
Acute basophilic leukemia
Acute basophilic leukemia is a leukemia at the expense of highly undifferentiated basophilic granulocytes. Sometimes it is the blast phase of Ph1-positive chronic myeloid leukemia, which has gone unnoticed.
It is characterized by the presence of a large number of blast cells in blood and bone marrow with a round or bilobed nucleus, with nucleoli, basophilic cytoplasm with basophilic granules, metachromatic, which can coexist with eosinophilic granules.

Acute panmyelosis with myelofibrosis
It is a very rare type of leukemia, which affects adults and rarely in children.
It can occur de novo or after treatment with alkylating agents or radiation therapy.
When there is a proliferation of a cell line that is accompanied by myelofibrosis, acute leukemia is diagnosed ≪with myelofibrosis≫; If blast proliferation is at the expense of different cell lines, it is usually classified as panmyelosis with myelofibrosis, a situation that in some cases may correspond to acute myeloblastic leukemia with myelodysplasia.

Myeloid sarcoma
It is a tumor of myeloblasts and myeloid cells in different maturation stages of extramedullary location or in bone.
Classically, it is known as chloroma because of its green color, frequent but not obligatory, due to a derivative of myeloperoxidase contained in cells. It has also been called myeloblastoma.
It affects 2-3% of LAM and 4-5% of chronic myeloid leukemias (CML). It can occur without evidence of acute leukemia in peripheral blood or bone marrow and precede its onset by months.
It can also be seen as a tissue manifestation of an already diagnosed LAM, and precede the blast transformation of a CML.
Any anatomical location can be affected, and the most frequent places are skin, lymph nodes, soft tissues on underlying bone, bone, periosteum, and viscera.

Acute leukemia of ambiguous lineage
Acute undifferentiated leukemias: morphology, cytochemistry, and immunophenotypic markers of blasts do not allow classifying them as myeloid or lymphoid.
Acute biphenotypic leukemias: blasts co-express myeloid and lymphoid antigens.
Bilinear leukemias: a myeloid cell clone coexists with a different one with lymphoid markers M1, M2, rarely M4 or M5. Never M3, M6, M7.


Cytogenetics
There is not a single chromosomal abnormality that is associated with this type of leukemias, but there is the presence of chromosomal abnormalities, with a high incidence of the Philadelphia chromosome.
Clinical features
They can affect adults and children, particularly those under 2 years of age. They can occur de novo or during a relapse after AML or ALL therapy.
The WBC count is often elevated, and many cases have a varying proportion of circulating blasts.
There is also no uniform criterion for how to treat these leukemias since there are not many reports.
It is considered a condition with a very poor prognosis for both adults and children.
Typical aml treatment (except M3)
Two phases:
Induction (induction of remission)
Consolidation (post-remission therapy)
Treatment needs to start as soon as possible after the diagnosis has been made as AML progresses rapidly.
Leucoestasis.
When a high number of leukemia cells are found, problems with circulation can occur. It takes a few days for chemotherapy to reduce the number of cells, so patients can undergo leukapheresis.
Induction
The purpose of the first part of the treatment is to remove as many leukemia cells as possible. The intensity of treatment depends on the age and general health of the patient.
In younger patients (under 60 years of age), induction consists of the use of two drugs: cytarabine (ara-C) and anthracyclines such as daunorubicin (daunomycin) or idarubicin. Sometimes a third drug, cladribine (leustatin, 2-CdA), is also given. Chemotherapy is given in a hospital and lasts about a week.
Patients with cardiac malfunction cannot be treated with anthracyclines, and must be treated with alternative drugs, such as fludarabine (Fludara) or topotecan.
In cases where the leukemia has spread to the brain or spinal cord, chemotherapy should be given into the cerebrospinal fluid. Radiation therapy can also be used.
Induction destroys most normal bone marrow cells as well as leukemia cells. Most patients develop dangerous pancytopenias, requiring antibiotics and transfusions (erythrocytes, platelets). As long as the low cell counts continues, the patient must remain in the hospital.
After treatment, the number of blasts in the bone marrow should be checked to confirm remission of the disease or if more chemotherapy should be given. Sometimes a stem cell transplant is recommended.
Induction of remission does not kill all leukemic cells, and a small number remain. Without consolidation treatment, the leukemia will return in a few months.
Consolidation (post-remission therapy)
Induction is considered successful if remission is achieved. More treatment is given to try to destroy any remaining leukemic cells (consolidation).
Young patients:
- Multiple cycles of high doses of ara-C
- Allogeneic stem cell transplantation
- Autologous transplant
- Ara-C is only used, in high doses, for 5 days every 4 weeks, for 3 or 4 cycles.
Elderly patients or patients in poor health cannot endure this consolidation treatment:
1 or 2 cycles of high doses of ara-C (lower than in young patients)
1 or 2 cycles of standard doses of ara-C, together with idarubicin, daunorubicin, mitoxantrone.
Non-myeloablative stem cell transplantation.
Management of patients with a favorable prognosis
Candidates for chemotherapy with ara-C. Administer increased doses of daunorubicin during induction and ara-C during consolidation.
The risk of relapse in these patients is not high enough to justify the risk of a stem cell transplant.
Desatinib: inhibits CKIT.

MicroRNA-193a represses c-kit expression and functions as a methylation-silenced tumor suppressor in acute myeloid leukemia
C-KIT: receptor tyrosine kinase
Management of patients with mk or unfavorable karyotypes without mk
They must undergo investigational drug therapies from induction.
They do not benefit from high doses of daunorubicin.
Nucleoside analogs:
Cladribine
Clofarabine or Clofarabine + low doses of ara-C.
Hypomethylating agents: azacitidine
Patients with AML and a mean age of 70 years: survival 24.5 months with azacitidine and 16 months with conventional therapy.
Patients with unfavorable cytogenetics: 12 months with azacitidine and 5 months with conventional therapy.
It must be used in combination with other drugs.
Stem cell transplantation with reduced intensity conditioning. Engraftment.
Remission of the disease
Complete morphological remission (CR): ANC ≥1,000 / ul, platelets ≥100,000 / ul, <5% blasts in bone marrow, without Auer rods, without evidence of infiltration in other tissues (There are no requirements for BM cellularity and concentration of Hb)
Complete morphological remission with incomplete cell count (CRi): Same as CR but with ANC <1,000 / ul and platelet count <100,000 / ul.
Partial remission: ANC ≥1,000 / ul, platelets >100,000 / ul, and at least a 50% decrease in bone marrow blasts or <5% with persistent Auer rods.

Clinical manifestations of acute lymphoblastic leukemias
Acute lymphoblastic leukemia represents 15-25% of all acute leukemias.
It is called adult when the patient exceeds the age of 15 years.
The physical examination shows, in half of the patients, lymphadenopathy, usually cervical, and splenomegaly.
Risk factors associated with CNS infiltration are considered:
The T phenotype
The L3 morphology of the FAB
A high baseline white blood cell count
96% of patients present a medullary infiltration of lymphoid blasts greater than 50% and dyshematopoietic morphological signs are not usually appreciated prior to any treatment in the normal residual cellularity.
Acute b-precursor lymphoblastic leukemia / lymphoblastic lymphoma
In the WHO classification, acute lymphoblastic leukemias (ALL) are included within the B and T-cell precursor neoplasms, and two entities are distinguished: B-precursor lymphoblastic leukemia/ lymphoma and T-precursor lymphoblastic leukemia/lymphoma.
B-Precursor lymphoblastic leukemia (B-ALL) and B lymphoblastic lymphoma (B-LL) are the same biological entity.
Both represent the proliferation of cells committed towards the B line, and show identical morphological, cytochemical and genetic characteristics, and only differ in the degree of bone marrow involvement.
Leukemias infiltrate the bone marrow and, generally, the peripheral blood.
Lymphomas can present primary nodal or extranodal involvement and, in the case of medullary infiltration, this is minimal or less than 25%; if it exceeds this figure, the term leukemia is more appropriate.
75% of ALL cases occur in children under 6 years of age, and 80-85% of cases are precursors of phenotype B.
Extramedullary involvement is common, with a predilection for the CNS, spleen, liver, lymph nodes, and gonads.

B-LL constitutes approximately 10% of lymphoblastic lymphoma cases, with a mean age of 20 years, it appears to be predominantly male, and the most frequently affected sites are skin, bone marrow, lymph nodes, and soft tissue.
Immunological classification is essential for the study of acute lymphoblastic leukemias, and is more important than merely morphological classification, since the immunophenotypic profile defines specific risk / prognosis subgroups.
Four subtypes of LAL-B are recognized, based on the greater or lesser differentiation of the proliferating clone: LALB1 or pro-B, LALB2 or common, LALB3 or pre-B and LALB4 or mature.

Acute t-precursor lymphoblastic leukemia/ lymphoblastic lymphoma
Leukemia: the spread of the disease reaches the bone marrow and peripheral blood.
Lymphoma: the process is confined to an extramedullary mass or the medullary infiltration is equal to or less than 25% of global cellularity.
The distinction is not always easy, and there may be exceptions.
They represent between 20 and 25% of ALL cases in adults, and 15% of children, and it is more common in men.
Patients with the T phenotype usually present elevated leukocyte counts, and with great frequency, lymphadenopathy and organomegaly.
T-ALLs constitute a heterogeneous group, as evidenced by the use of monoclonal antibodies and molecular biology techniques.
T-ALL and T-LL can be stratified according to the different stages of intrathymic differentiation, according to the number and sequence of the antigens expressed: CD3 cytoplasm, CD2 and CD7, the earliest, followed by CD5 and CD1a and, finally, by membrane CD3.


Typical treatment of all
Long-term chemotherapy.
Three phases:
Induction (or induction of remission)
Consolidation (intensification)
Maintenance
Total treatment takes approximately 2 years, with the maintenance phase taking most of it. The intensity of treatment depends on the ALL subtype and other prognostic factors.
ALL can spread to the area around the brain and spinal fluid. Sometimes this has already happened when ALL is diagnosed.
An important part of the treatment of ALL is the prophylaxis of the nervous system since sometimes the study of the spinal fluid does not show leukemic cells but it does not mean that they are not there, they can be in very small quantities.
Induction
The goal is remission: bone marrow without leukemia cells, with normal cells and counts. It is not necessarily a cure as the leukemia cells can still remain.
It is an intense chemotherapy phase that lasts for about a month. Different combinations of drugs are used:
Vincristine
Dexamethasone or prednisone
Doxorubicin (Adriamycin), daunorubicin, or anthracycline-like drugs.
Based on the prognostic factors of the patients, some regimens may include:
Cyclophosphamide
L-asparaginase
Etoposide (VP-16)
High doses of methotrexate or ara-C
Imatinib (Gleevec) for ALL with the Philadelphia chromosome.
Antibiotics, transfusions.
Cns prophylaxis
Treatment to prevent leukemia cells from spreading to the CNS (prophylaxis) is similar to that used to treat leukemia that has already spread.
It often begins with induction and continues into the other phases of treatment.
It may include one or more of the following:
Chemotherapy injected directly into the spinal fluid (intrathecal chemotherapy); methotrexate, arac-C, prednisone. Lumbar injection or through the Ommaya resevoire.
High-dose intravenous methotrexate or ara-C.
Radiation therapy to the brain or spinal cord.

Consolidation (intensification)
If the leukemia goes into remission, the next phase consists of another short course of chemotherapy, using many of the drugs that were used in induction. It lasts for about a few months.
They are given in high doses. Continuation of CNS prophylaxis.
Imatinib.
Allogeneic or autologous stem cell transplantation.
Maintenance
After consolidation, patients generally undergo a maintenance chemotherapy program with methotrexate and 6-mercaptopurine.
In some cases it can be combined with vincristine and prednisone.
Imitanib.
Maintenance usually lasts two years, and CNS prophylaxis must be maintained.
Prognostic
Complete remission
ALL: 90%
AML: 60% to 70%
5-year disease-free
ALL: 60%
AML: 20%
Conclusion
Acute myeloid leukemia has a worst prognostic when It has evolved from another hematological disorder like a myelodysplastic or myeloproliferative syndrome.
The monosomic Karyotype also has a worst prognostic and still has not good treatment. Acute lymphoid leukemia needs three treatment stages and most of the times the CNS has already been invaded.