
Hemolytic anemia
Introduction
Hello, welcome to this 8th class of the Hematology course provided by the University of Guanajuato. In this class we are going to review some basic concepts of hemolytic anemia. Erythrocytes normally survive around 120 days within the circulation, and they are destroyed by the mononuclear phagocytic system. When hemolysis occur, erythrocytes are destroyed within a very short time they were released from the bone marrow, due to alterations inside and outside the erythrocyte. Normally, bone marrow can mitigate the erythrocyte destruction rate but when It Is greatly Increased, the hemoglobin levels are decreased, reticulocytes and erythropoietin levels are Increased, as well as some other metabolic parameters, like bilirubin.
Erythrocyte’s alterations could be structural, enzymatic, hemoglobin synthesis, or external, among others. Each one has Its own characteristics, which will be reviewed In this class, as well as some treatment options. So, let’s begin with this Interesting subject, hope will help you better understand this subject.
Content development
Hemolysis Is defined as a premature destruction of red blood cells.
The common denominator: lesion of the erythrocyte that conditions its early disappearance from circulation.
From leaving the bone marrow until they are eliminated by the phagocytic mononuclear system (SMF), erythrocytes live for approximately 120 days, and when this period decreases, there is hemolysis.

Decreased hemoglobin concentration (anemia) tends to be counteracted by erythropoiesis stimulus to an increase in erythropoietin.
Compensated hemolysis: the existence of an increase in the concentration of reticulocytes (reticulocytosis).
Typical bone marrow can increase its erythrocyte production sic to eight times than usual; there must be significant destruction for anemia to occur.

Hemolytic anemia occurs when bone marrow activity cannot compensate for the loss of red blood cells.
The severity of anemia depends on the onset of hemolysis, whether gradual or abrupt and the extent of red blood cell destruction.
Moderate hemolysis can be asymptomatic, while severe hemolysis can cause angina or pulmonary decompensation and even be fatal.

Classification of hemolytic anemias
Intracorpuscular: intrinsic erythrocyte defect, inherited or acquired.
Extracorpuscular: secondary lesion of the erythrocyte by agents of plasma or vascular origin.

Intravascular hemolysis is the destruction of erythrocytes in the circulation with the release of the contents in the plasma.
The most common extravascular hemolysis is removing and destroying membrane-altered erythrocytes by macrophages in the spleen and liver.

SRE: Endothelial Reticulum System
All hemolytic processes always have a mixed behavior, when the extravascular mechanism predominates, the fundamental characteristic is an increase in iron in the SMF and the plasma concentration of bilirubin (jaundice), a pigment resulting from the degradation of the heme group.
Normal erythrocyte catabolism
90% extravascular hemolysis
Spleen, liver, bone marrow, lymph nodes
Globin + Heme
Iron to transferrin, globin to amino acids (pool)
Heme + Heme oxygenase: Biliverdin + CO2
Biliverdin is reduced to unconjugated bilirubin that binds to albumin and is transported to the liver
In the liver it dissociates from albumin and is conjugated with glucuronic acid (conjugated bilirubin)
Conjugated bilirubin: direct, water soluble
Unconjugated bilirubin: indirect, insoluble in water, reacts with the addition of alcohol.

Conjugated bilirubin is excreted with the bile in the intestines and is converted to urobilinogen (feces)
A small portion is reabsorbed and goes to the liver; a small% is excreted in the urine.

Extravascular hemolysis (hemolytic disorder)
Erythrocytes live 15-20 days.
Bone marrow: compensated hemolytic status.
Increased unconjugated bilirubin in serum and urobilinogen in urine.

Intravascular hemolysis
Free hemoglobin in plasma
Immediately bound and eliminated by haptoglobin (Hpt), a plasma protein synthesized by the liver.
Suppose it exceeds the binding capacity of Hpt. In that case, the excess hemoglobin is eliminated directly by the kidney. It is taken up by the tubular cells, which degrade it and accumulate iron in the form of hemosiderin.
When intravascular hemolysis is very intense and exceeds the capacity of the kidney tubular cells to fixate, hemoglobin, already degraded to methemalbumin and other compounds, is eliminated in the urine (hemoglobinuria), which, as a result, acquires a dark color very characteristic and similar to that of ≪Coca-Cola.≫

When intravascular lysis of erythrocytes occurs, hemoglobin is withdrawn from the circulation by various mechanisms:
- The first option is to use circulating haptoglobin with the formation of the haptoglobin-hemoglobin complex. This complex bypass the kidney filter and is eventually taken up by hepatocytes, where hemoglobin is separated from haptoglobin and metabolized to bilirubin.
- The second option, which is less important than the first, consists of the formation of 2 complexes in the circulating blood. There are globin separation and binding of heme to plasma hemopexin. In cases of severe intravascular hemolysis, there is the formation of meta-heme-albumin. These complexes also do not pass the kidney filter and are metabolized in the liver.
- The third option is that the free hemoglobin – formed in excess – passes through the glomeruli, being then partially taken up by the proximal tubules of the kidney until they are saturated. In the renal tubules, hemoglobin is unfolded with the formation of hemosiderin and ferritin, which are reabsorbed into the general circulation. The rest of the hemoglobin, which the renal tubules could not take up, is excreted with the urine (hemoglobinuria).

Clinical classification
Acute
Appears abruptly in a previously healthy subject.
It is characterized by a quickly decrease in hemoglobin concentration, striking clinical features, and occasionally hemoglobinuria (dark urine).
In the case of a child or young adult, a history of drug intake is highly suggestive of glucose-6-phosphate dehydrogenase (G6PD) deficiency, drug-induced hemolytic anemia, or unstable hemoglobinopathy.
On the contrary, in an adult subject, it is most likely that it is hemolytic anemia of immune origin (autoimmune or immune medical hemolytic anemia), microangiopathic (mechanical hemolysis) or paroxysmal nocturnal hemoglobinuria (PNH).
Chronic
It appears slowly, with a variable decrease in hemoglobin concentration and clinical signs of chronic hemolysis, such as jaundice and splenomegaly.
The intensity of jaundice and its distribution (skin and/or mucosa) is variable, although in most cases, it is limited to the ocular conjunctiva (conjunctival sub jaundice).
Hemolytic jaundice is due to excess unconjugated or free bilirubin (also called ≪indirect≫), so, even when it is very intense, it is never accompanied by choluria (acholuric jaundice) and pruritus, except when it coexists with a hepatobiliary disease (lithiasis and bile duct obstruction).

Genetic classification
Hereditary: defects in the genes that encode the synthesis of proteins essential for the life of the erythrocyte and that are transmitted with hemoglobin: hemoglobin, membrane proteins, and enzymes.
In most cases, they appear shortly after birth (congenital) and remain throughout life.
Acquired: they appear in the course of life and are not transmitted with heredity, but like these, they are due to alterations of the erythrocyte induced by very different causes (some of them also genetic), reduce their survival in circulation.
Diagnosis of hemolithic syndrome
Demonstration of the existence of hemolysis
Performing various general laboratory tests that allow to objectify, albeit indirectly, the early elimination of circulating erythrocytes.

The most characteristic laboratory finding of hemolysis is reticulocytosis.

Label the patient’s erythrocytes with radioactive chromium (i1Cr) and re-inject them, proceeding then to practice extractions serial (every 3 or 4 days for a minimum of 20 days) and measure, each time, the intensity of radioactivity residual.
Reports on the survival of red blood cells in the circulation and, at the same time, quantifies the intensity with which said elimination is carried out.


Hereditary spherocytosis
Autosomal hereditary defect, the erythrocytic cytoskeleton proteins are affected and these cells are destroyed by the spleen causing a chronic hemolytic anemia. The mean corpuscular hemoglobin Is usually Increased.
Splenectomy can decrease extravascular hemolysis and prevent long-term complications such as cholelithiasis and aplastic crisis. Due to the inherent risk of infections and sepsis, splenectomy is recommended in patients older than five years old.


Hereditary eliptocytosis
Mutation in the genes that encode the spectrins of the cytoskeleton.

Due to their flexible skeleton, normal erythrocytes can deform when passing through small-caliber vessels, later returning to their standard shape.
In hereditary elliptocytosis, the deformed red blood cell becomes fixed in the elongated shape as the membranous skeleton reorganizes.

Paroxysmal Nocturnal Hemoglobinuria (PNH)
It is considered hemolytic anemia but is a bone marrow myeloproliferative disorder with intravascular hemolytic anemia due to the increased susceptibility of the red blood cell to complement.
Defect present in erythrocytes, platelets, and granulocytes.
Hemolytic disorder acquired by an abnormality in the red cell membrane.
Clonal and acquired disease caused by a somatic mutation in the PIG-A gene that encodes a protein involved in the synthesis of Glycosylphosphatidylinositol (GPI), which serves as an anchor for many proteins in the cell membrane.
The mutation occurs in the hematopoietic stem cell. It leads to a partial or total deficiency of the PIG-A protein with the consequent alteration in the synthesis of the anchor GPI.
The result of the loss of these proteins from the cell surface increases sensitivity to complement-mediated cell destruction.

The cause of hemoglobinemia during sleep is still under discussion. Still, it is believed that it is the decrease in the pH of the blood during sleep that facilitates the fixation of complement to the PNH cells.
Other causes: infections, vaccines, transfusions (increase the amount of complement available).
It can progress to acute non-lymphocytic leukemia (1 to 3%). Patients are predisposed to intravascular thrombosis (portal or venous circulation of the brain).

Erythrocyte enzyme defects
The maturation process of erythroblasts involves the disappearance of all specific metabolic pathways in cells, leaving only the essential components to maintain energy supply and defense against oxidizing agents.
To carry out the gas exchange, erythrocytes need three metabolic pathways.
- Anaerobic glycolysis: ATP production (maintenance of cell structure and function)
- Antioxidant pathways: synthesis of glutathione (GSH), maintenance of the oxidation state of Fe from hemoglobin
- Nucleotide metabolism: maintenance of nucleotides (purines and pyrimidines).
Production of 2,3-diphosphoglycerate: affinity of hemoglobin for oxygen.
Erythro-enzymopathies associated with hemolytic anemia
The degree of hemolysis depends on the severity of the enzyme deficiency, a consequence of the properties of the mutant enzyme concerning functional abnormalities, molecular instability, or both.
In addition to anemia, patients present with jaundice (increased bilirubin), reticulocytosis, and splenomegaly.
The morphology of the erythrocytes is observed with anisocytosis and polychromasia, without any specific changes except for echinocytes and basophilic stippling.
Spherocytosis and elliptocytosis (non-spherocytic hemolytic anemias) are not observed.
Favism: It is an intolerance to the ingestion of beans or the inhalation of the Vicia faba plant native to Asia from which this legume comes.
The compounds Vicina and Convicina act as glutathione oxidants (coupled with the deficiency of G6PD activity, being responsible for maintaining low levels of reduced glutathione.

Pyruvate Kinase (PK) deficiency
Catalyzes the irreversible transfer of a phosphate group from phosphoenolpyruvate to ADP to form pyruvate and ATP (glycolytic pathway)
Causes a deficiency in ATP production.

Pyrimidine 5 nucleotidase deficiency
Involved in the catabolism of the RNA of young erythrocytes (reticulocytes), it must degrade this molecule as a final step in the maturation of erythrocytes. Nucleotides are not used by erythrocytes and must be exported from the cell.
Autosomal recessive disorder characterized by chronic hemolytic anemia associated with basophil stippling (RNA precipitation) due to accumulation of high concentrations of pyrimidine nucleotides within the erythrocyte.
Also related to anemia due to lead poisoning and thalassemia.

Hemoglobinopathies
Chronic hemolysis can be due to disorders of hemoglobin synthesis, including sickle cell anemia and thalassemias.
Thalassemias are a heterogeneous group of anemias characterized by defects in synthesizing the alpha or beta subunits of the hemoglobin tetramer (a2b2).
The deficiency in a globin chain causes a decrease in hemoglobin and its intracellular precipitation from the excess chain, damaging the membrane causing hemolysis.
There are two forms: alpha (hemoglobin H disease) and beta-thalassemia (intermediate and major).

Normally, people have 4 genes for alpha globin.
People with alpha thalassemia may be missing 1, 2, 3, or 4 globin genes.
Deletion T Point mutation
The most frequent mutations are deletions.

Alpha thalassemia trait
Individual with two abnormal alpha-globin genes
Abnormal genes can be on the same chromosome (cis position), or they can be on each of the chromosomes (trans position).
It May be confused with iron deficiency anemia (small red blood cells).
Hemoglobin H
Anemia is moderate to severe and can lead to serious health problems such as hepatosplenomegaly, jaundice, bone deformities (overgrowth of the jaw and forehead), and fatigue.
This hemoglobin is unstable and forms inclusion bodies (Heinz bodies) that damage the red blood cell membrane.
It has a high affinity for oxygen but does not present the Bohr effect or heme-heme interaction, so the gas exchange becomes inefficient under physiological conditions.
Red blood cells are susceptible to oxidative stress.
Hydrops fetalis: It is characterized by causing severe edema, swelling in the fetus or newborn due to an excessive amount of fluid that leaves the bloodstream and enters various body tissues. Intrauterine transfusion stops and even reverses the pathophysiological process that causes anemia, which can progress to hydrops. It can also decrease the number of exchange transfusions in the immediate neonatal period.
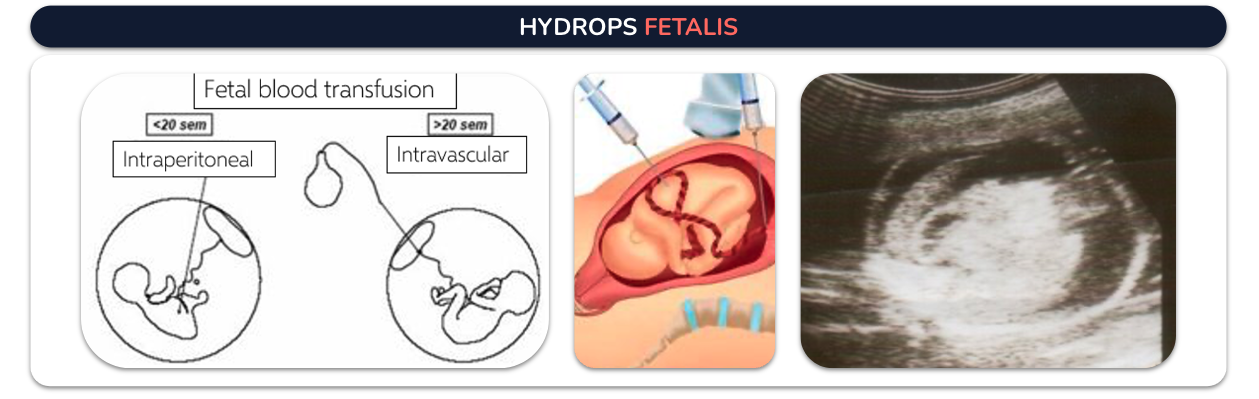
Beta thalasemia
Classification:
Thalassemia major: Cooley’s anemia (first 2 years of life)
Thalassemia intermedia
Thalassemia minor In contrast to alpha thalassemias, the most common molecular abnormalities are point mutations and insertions or deletions limited to a few nucleotides.

Thalassemia major
Genotype: homozygous 8 βº.
Individuals with thalassemia major usually present with severe anemia within the first two years of life.
In untreated or poorly transfused patients:
- Stunted growth
- Hepatosplenomegaly
- Development of masses due to extramedullary hematopoiesis
- Skeletal changes
Thalassemia major

They require regular GR transfusions.
This method results in iron overload.
In some cases, the spleen needs to be removed.
Marrow transplantation is the only possible definitive cure.
Thalassemia intermedia
Thalassemia intermedia begins to manifest later, with moderate anemia.
The main clinical symptoms are erythrocyte marrow hypertrophy with medullary and extramedullary hematopoiesis with their respective complications:
- Deformed bones
- Typical facial changes
- Gallstones
- Leg ulcers
- Greater predisposition to thrombosis.
Treatments:
May require splenectomy
Folic acid supplementation
Treatment of extramedullary erythropoietic masses
Leg ulcers
Thalassemia minor
Thalassemia minor is clinically asymptomatic, but some patients usually have mild anemia.
Clinical Diagnosis
- Thalassemia major: in children under two years of age, severe microcytic anemia, mild jaundice, and hepatosplenomegaly
- Thalassemia intermedia: in older children with similar clinical pictures.
- Thalassemia minor: In general, they are asymptomatic and may present with very mild anemia.
Hematological diagnosis

Peripheral blood smear
Affected individuals show morphological changes in GR (microcytosis, hypochromia, anisocytosis with poikilocystosis)
The number of erythroblasts is related to the degree of anemia and increases markedly after splenectomy.
Qualitative and quantitative analysis of Hb
Determination of Hb concentration in whole blood, using the cyanomethaneHb method
Cellogel electrophoresis: indicates the amount and type of Hb present.
The Hb pattern in β-thalassemia varies:
Homozygous β0 thalassemia (major):
- HbA is absent
- HbF constitutes 92-95% of total Hb. .
β + homozygous and β + / β0 heterozygous:
- HbA are between 10 and 30%
- HbF between 70-90%.
- HbA2 is variable in homozygous BT and is improved in beta thalassemia minor.
Treatment:
Transfusions:
Hb values fall below 7gr / dl
Facial deformations
Poor growth
Bone expansion
Increased splenomegaly
Treatment of Fe overload
Splenectomy
Chelation therapy:
Administer Iron chelators for elimination in urine and feces (from 10 to 20 transfusions or at ferritin levels above 1000 ng/ml).
Acquired hemolytic anemias
MICROANGIOPATHIC ANEMIA
Fragmentation of red blood cells in the circulation leading to intravascular hemolysis.
Passage of erythrocytes through fibrin deposits present within the lumen of arterioles and capillaries or through the damaged epithelium and vessel walls.
Disseminated intravascular coagulation
Thrombo-hemorrhagic disorder
Excess thrombin activation and fibrin deposition.
Blood poisoning (gram negative bacteria).
Eclampsia, Amniotic fluid clots.
Cancer
Extensive burns
Transfusion reactions
Viper venom

Thrombotic Thrombocytopenic Purpura
It belongs to the group of microangiopathic hemolytic anemias.
Erythrocyte fragmentation and platelet consumption.
A Heterogeneous clinical syndrome characterized by:
Hemolytic anemia with red blood cell fragmentation
Thrombocytopenia
Variable neurological dysfunction
Fever
Progressive kidney failure
Thrombotic occlusions disseminated in the microcirculation.
Immune diseases, infections, pregnancy, hereditary factors.
Unlike DIC, the common complication is hemolytic anemia with fragmented red blood cells.
Symptoms are the result of platelet thrombi in small vessels leading to infarcts and minor bleeding.
Treatment: plasma exchange is generally associated with steroids; 10% of patients who receive this treatment may be resistant to it, and 50% of them present with exacerbation of the clinical picture.
In cases resistant to this treatment, some reports suggest administering rituximab.
Immune hemolytic anemia
Result of an immune event that occurs on the surface of the erythrocyte
Direct: Action of an antibody on the membrane with or without complement
Indirect: an immune complex that is adsorbed on the erythrocyte and fixes complement, which is generated by its lysis by the action of macrophages
Autoimmune hemolytic anemia
Autoantibodies

The direct antiglobulin test, better known as direct Coombs, demonstrates the presence of antibodies or complement on the surface of the red blood cell and is a cornerstone in the diagnosis of autoimmune hemolytic anemia.

Drug hemolytic anemia
- Hapten: does not fix complement, extravascular hemolysis.
- Immunocomplexes: adsorbed on the erythrocyte membrane, activates complement, intravascular hemolysis.
- Autoantibodies.

Hemolytic transfusion reactions and hemolytic disease of the newborn.

Conclusion
Hemolytic anemia occurs when the bone marrow cannot compensate the destruction of erythrocytes by any alteration and hemoglobin levels dramatically decrease.
Alterations in erythrocytes can be intrinsic erythrocyte defect, inherited or acquired and a secondary lesion of the erythrocyte by agents of plasma or vascular origin.
Erythrocyte’s destruction causes reticulocytes, erythropoietin and non-conjugated bilirubin levels Increase.